Management of Advanced Cutaneous Melanoma
Event
DC Resident Consortium Lecture
Overview & Learning Objectives
The lecture on March 10th has the following objectives:
- We will review the work up for lesions suspicious for melanoma
- Understand the current therapeutic landscape for high-risk localized cutaneous melanoma
- Review treatment strategies for high-risk regionally metastatic melanoma
- Discuss management options for patients with metastatic melanoma
Here, we provide a primer for that lecture.
First we start with a section reviewing the molecular mechanisms important in melanomagenesis.
Then, we have four clinical cases that are provided so that you can think through them ahead of the lecture. We will go through these cases as a group on Thursday.
Molecular Mechanisms of Melanomagenesis
Questions
- Prior to reading this section, ask yourself the following questions:
- What are the most common driver mutations in melanoma?
- Which oncogenic proteins are targeted by FDA-approved therapies?
- If the majority of nevi have BRAF V600 mutations (and they do), why don’t all nevi eventually transform into melanoma?
- What are the most common driver mutations in melanoma?
Signaling pathways in melanoma
Melanoma results from the dysregulation of both cell autonomous and non-autonomous elements. Cell autonomous factors include aberrations in a number of cellular circuits including the phosphatidylinositide 3-kinase (PI3K) pathway, telomerase promoter, the retinoblastoma pathway, and the mitogen-activated protein kinase (MAPK) pathway (Figure 1). We will provide a basic overview of them here (this is meant to be a primer and not a comprehensive review).
Oncogene-targeted therapy became a possibility following the 2002 discovery that activating mutations of the proto-oncogene v-Raf murine sarcoma viral oncogene homolog B (BRAF) drive nearly half of melanomas (Davies et al., 2002). BRAF, a serine-threonine kinase in the MAPK pathway, is normally activated by the proto-oncogene RAS. Activation of BRAF by RAS results in signal transduction via the phosphorylation of the mitogen-activated extracellular signal-regulated kinase (MEK). Pathogenic mutations in BRAF - most commonly V600E, but occasionally other substitutions such as V600K and others - result in constitutive activation of the kinase domain. Consequently, RAS-independent hyperactivation of downstream mediators such as MEK and extracellular signal-regulated kinase (ERK) can occur.
Enhanced activity of this pathway can lead to cell-cycle dysregulation (which we’ll discuss more further in the post) and resistance to apoptosis. The discovery that gain-of-function alterations in BRAF were a key determinant in melanomagenesis drove the development of therapeutic strategies to intervene at multiple nodes in the MAPK pathway.1
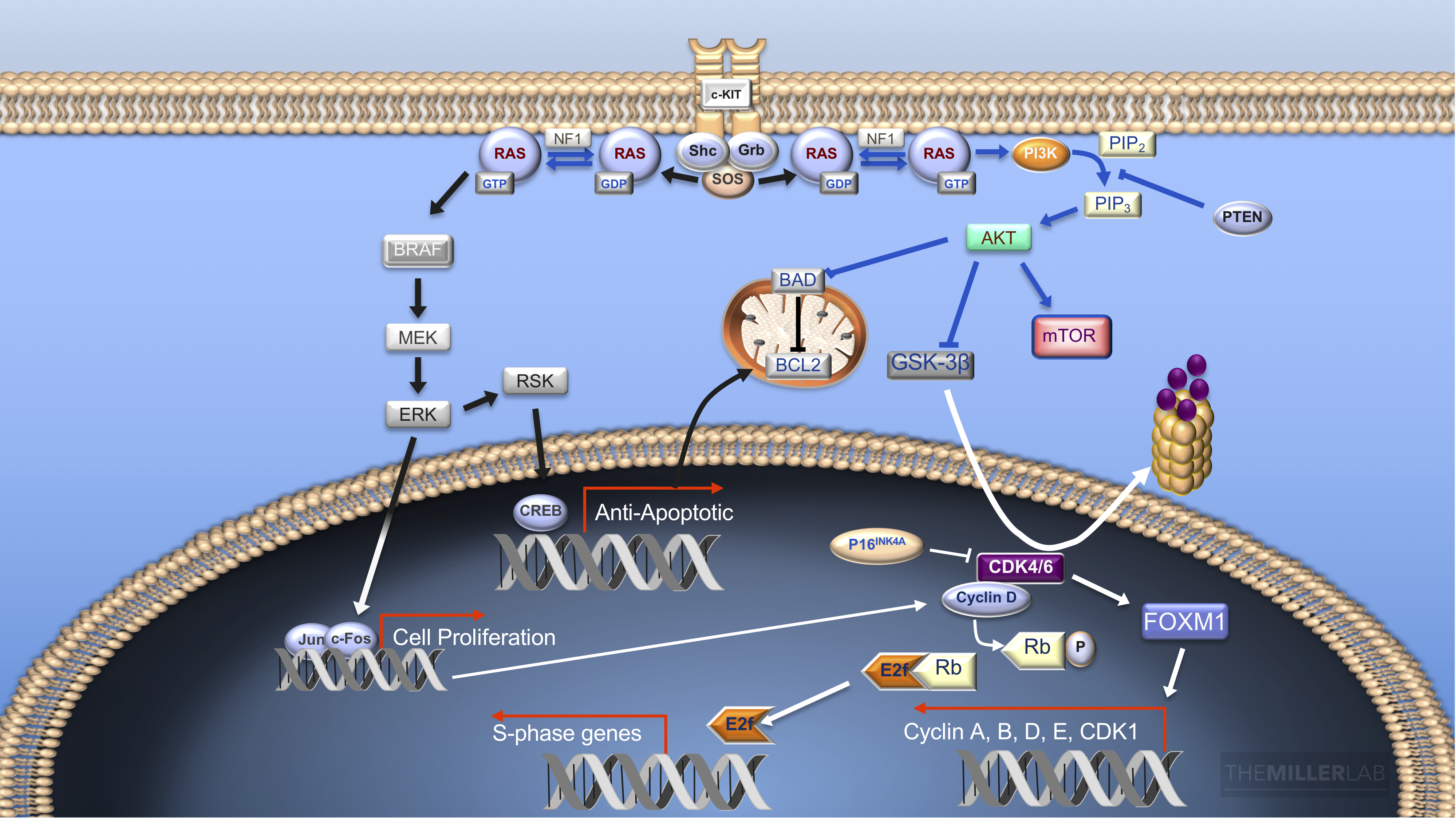
Figure 1: Signaling Pathways in Melanoma. Stimuli from mitogenic receptors (e.g.c-KIT) results in activation of the proto-oncogene RAS. Activated RAS promotes cell survival and growth by triggering output of both the MAPK kinase pathway (BRAF-MEK-ERK) as well as the PI3K-PIP3-AKT pathway. Cellular growth is promoted by transcription of cyclin D family members via MAPK and PI3K signaling. MAP kinase signaling promotes cyclin D1 expression, via the proteins JNK, ERK, Jun and c-fos, leading to the liberation of E2F from RB. Cyclin D transcription is also triggered when activated AKT stimulates the mammalian target of rapamycin (mTOR) to inhibit a suppressor of eIF4E, 4E-BP1. Furthermore, cyclin D–CDK4/6 complexes lead to cellular proliferation through multi-site phosphorylation of FOXM1, which leads to the expression of mitogenic genes (e.g. CDK1 and members of the cyclin A, B, D and E families). Several anti-apoptotic outputs are also induced downstream of RAS. Upregulation of BCL2, via RSK and CREB, antagonizes apoptosis at the level of the mitochondria. BCL2 activity is further enhanced by the AKT-mediated phosphorylation of the pro-apoptotic BAD. Downregulation of RAS occurs via GTPase-activating proteins such as NF1. These facilitate the hydrolysis of GTP to GDP, effectively turn RAS “off”. On the other hand, GEFs stimulate the release of GDP, allowing RAS to bind GTP, leading to its activation.1
Cell-Cycle and Melanoma Oncogenesis
Cell growth is regulated by a complex and dynamic interplay between signals that govern cell metabolism, differentiation, proliferation and cell death. Key in this process is the “cell cycle”. A basic understanding of the cell cycle can enhance your understanding of key components of melanoma pathogenesis, inherited cancer syndromes and ongoing drug-development programs (this is indeed an active area of investigation in melanoma). The cell cycle incorporates three checkpoints (G1, G2, and M) to ensure controlled DNA replication and cellular proliferation. Disruptions in the integrity of these checkpoints can result in inappropriate proliferation, genomic instability and cellular transformation (Figure 2).
An intricate relationship of three families of proteins - cyclins, cyclin-dependent kinases and cyclin-dependent kinase inhibitors (CDKi) - regulates these checkpoints and governs the cell cycle. The cell cycle is utilizes cyclins of four classes (A, B, D and E), three interphase CDKs (CDK2, CDK4 and CDK6) and a mitotic CDK (CDK1) (Malumbres et al., 2009). Cell cycle commitment occurs in G1; critical to this checkpoint is the interactions between cyclin-CDK complexes and members of the retinoblastoma (RB) tumor suppressor gene family. After receiving mitogenic stimuli, quiescent cells are released from G0 as members of the D-type cyclins are expressed and activate CDK4 and CDK6. These cyclin D-CDK4/6 complexes begin the initial phosphorylation of RB family member proteins during G1 (Figure 2). Following initial phosphorylation, the cycle E-CDK2 complex phosphorylates RB further, liberating the transcription factor E2F from RB. This results in the activation of a gene expression program required for DNA replication. Prior to this critical junction (known as the “restriction point” (Pardee, 1974)), progression of the cell cycle is reversible and dependent on mitogenic stimuli.
Past the “restriction point”, cells are committed to the cell cycle and extracellular signals are not necessary for further growth. Following the “restriction point”, RB is maintained in a phosphorylated state by cyclin A-CDK2 complexes and the mitotic CDK – CDK1.1
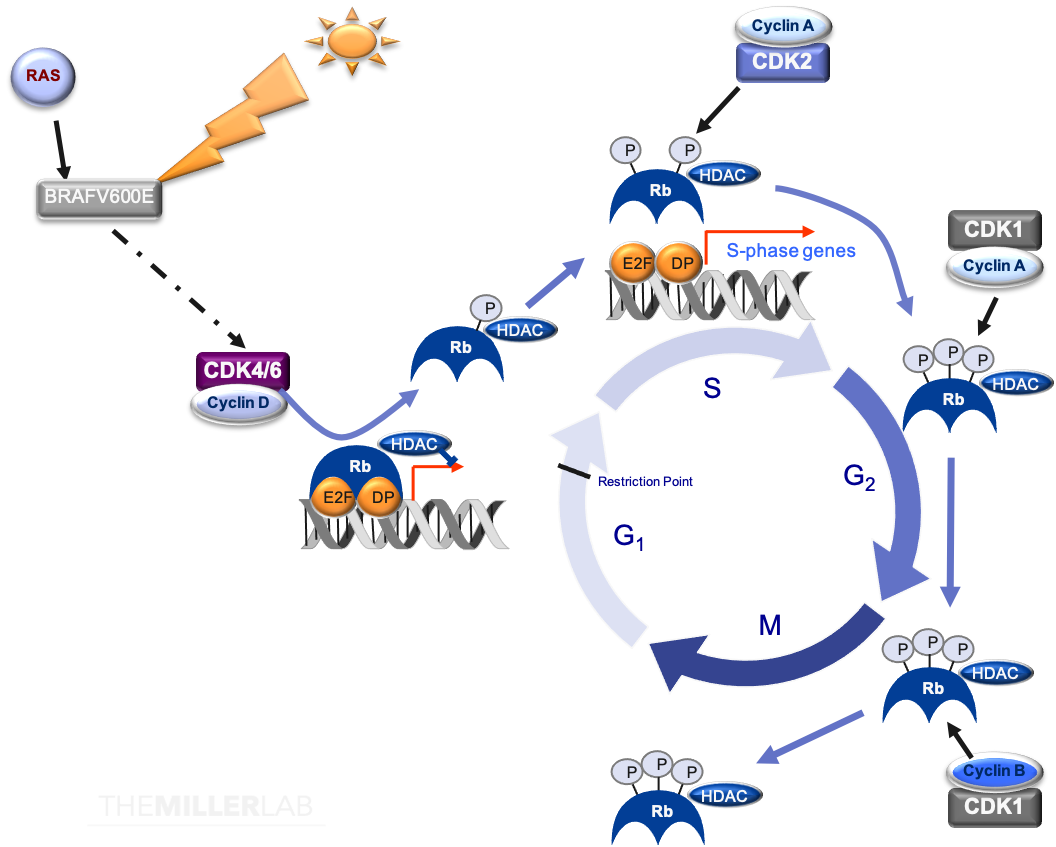
Figure 2: The Cell Cycle. The cell cycle is governed via the interactions of highly-regulated proteins designed to maximize genomic fidelity. Cells enter the cell cycle following the reception of mitogenic signals (e.g. from stimuli such as RAS activation). This can lead to the activation of the cyclin D-dependent kinases, CDK4 and CDK6, by way of increased production of cyclin D. This then triggers the release of CDK4/6 from inhibitory INK4 and CIP/KIP proteins (e.g. p15/p16 and p21CIP1/p27KIP1, respectively). Early in G1, RB is initially phosphorylated by cyclin D–CDK4/6 complexes, effectively abrogating the RB-dependent inhibition of the transcription factor E2F. E2F is then released from the partially phosphorylated RB, which allows it to promote transcription of genes that are required to progress through the G1/S checkpoint. RB is further phosphorylated in late G1 by cyclin E–CDK2 complexes, enabling E2F transcription factors to express genes necessary for DNA synthesis. Following the entry into S phase, a hyperphosphorylated state of RB is perpetuated by the actions of group of cyclin-CDK complexes (e.g. cyclin A–CDK2, cyclin A–CDK1 and cyclin B–CDK1). Upon the conclusion of mitosis, RB is dephosphorylated by phosphatases allowing recombination with E2F and DP to prevent unregulated cell proliferation.1
Cellular Senescence and Melanomagenesis
Although activation of the MAPK pathway is critical in the transformation of melanocytes, it is not sufficient for oncogenesis. Dysregulations in cell-cycle checkpoints and cellular senescence are necessary. For example, the vast majority of nevi have alterations in the MAPK pathway (e.g. BRAF V600 mutations); however, as we know, most nevi never undergo malignant transformation. This is in part due to the fact that unregulated and constitutive activation of the MAPK pathway often triggers a protective mechanism, called oncogene-induced senescence (Figure 3). Thus, the majority of melanomas have an aberration of some element of the p16INK4A:cyclin D-CDK4/6:RB pathway, in addition to abnormalities of the MAPK pathway (Bennett, 2008). Not surprisingly, therapies that are also targeting proteins in the cell cycle (e.g. CDK4) are being investigated actively for patients with melanoma.
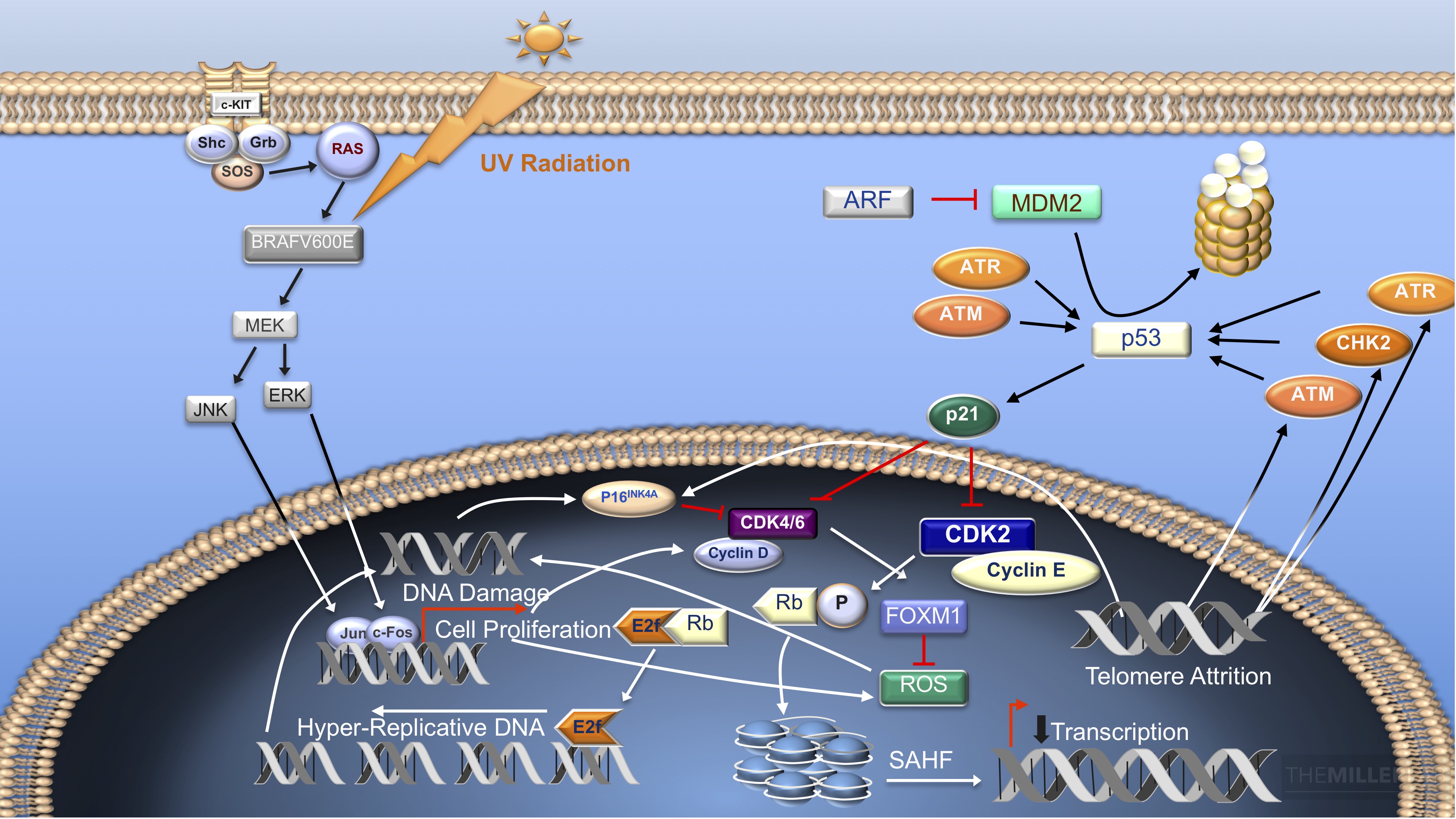
Figure 3: Oncogene-Induced Senescence: Cellular senescence can be initiated via several pathways that incorporate common effectors. While the mechanisms governing the cellular senescence program have yet to be fully elucidated, telomere shortening is thought to trigger cellular arrest by triggering INK4a-RB signaling and the p53-p21 pathway via molecules such as ATM, ATR and CHK2. Inactivation of CDKs can also occur secondary to activation of the DNA damage response (DDR). The DDR can be activated by a variety of genotoxic stimuli including reactive oxygen species (ROS), hyper-replicative DNA and ultraviolet radiation. The DNA damage response stimulates INK4a-mediated inhibition of CDK4/6 and the p53-p21 pathway in part via ARF inhibition of MDM2-mediated ubiquitination of p53 and activation of ATM. Activation of INK4a inhibits the CDK4/ 6-mediated activation of FOXM1, which antagonizes ROS-mediated DNA damage. The senescence cascade is also activated in certain contexts of persistent mitogenic stimulation, such as constitutive activation of RAS or RAF proteins (referred to as oncogene-induced senescence (OIS) (Serrano et al., 1997). OIS involves p38 MAPK and activates RB and p53 via HBP1 and PRAK, respectively (Funayama and Ishikawa, 2007; Wang et al., 2002). Active RB contributes to formation of senescence-associated heterochromatin foci (SAHF), and this heterochromatization of DNA precludes transcription of E2F target genes, contributing to the overall state of irreversible cell-cycle arrest.1
Clinical Cases
CASE 1
Synopsis
- 71 year old Fitzpatrick skin type 2 male with no significant PMH presents with this lesion on his chest x 3 months
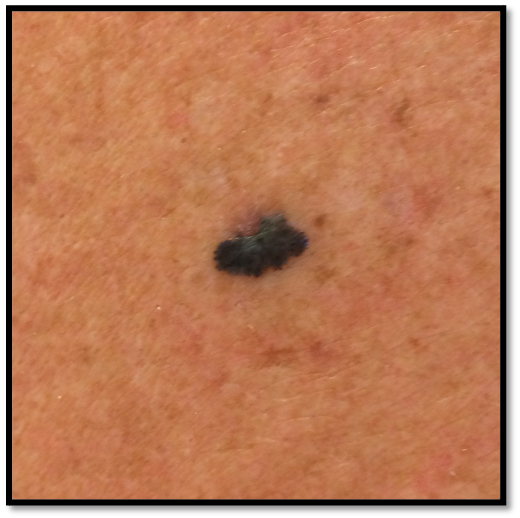
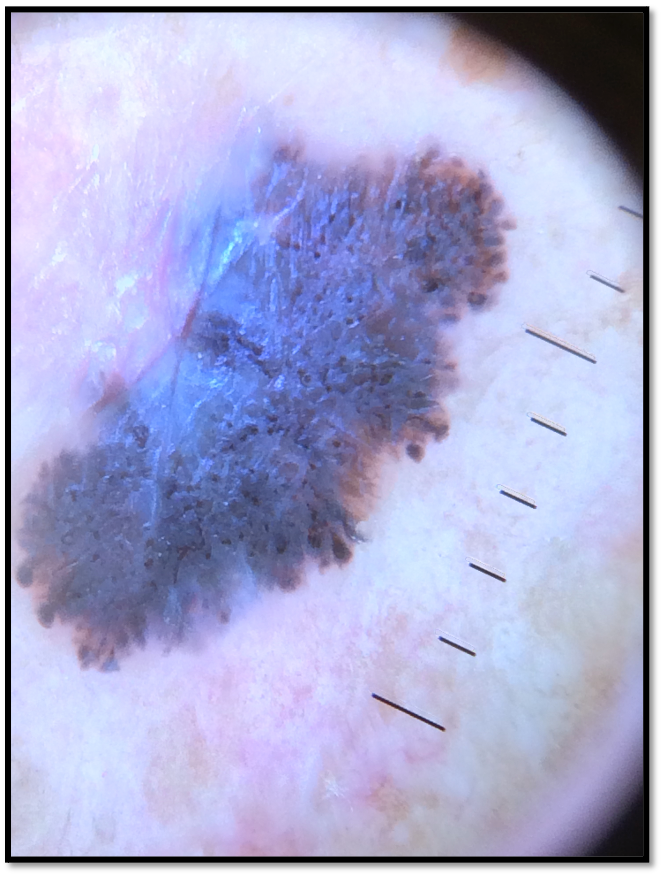
Case 1 Clinical Questions:
- What technique(s) would you use to diagnose this lesion?
A biopsy was performed.
Pathology reveals:
- HISTOLOGIC TYPE: Superficial spreading melanoma
- PRECURSOR LESION: Not identified
- MAXIMUM TUMOR THICKNESS: 3.0 mm
- ANATOMIC LEVEL: At least level IV
- ULCERATION: Present
- MITOTIC RATE: 9 per mm2
- LYMPHOVASCULAR INVASION: Present, Foci suspicious for lymphovascular invasion
- METHOD OF DETECTION: Hematoxylin and eosin
- RADIAL GROWTH PHASE: Present
- VERTICAL GROWTH PHASE: Present
- TYPE OF VERTICAL GROWTH: Epithelioid
- MICROSATELLITES: Cannot be assessed
- PERINEURAL INVASION: Cannot be assessed
- TUMOR-INFILTRATING LYMPHOCYTES: Present, nonbrisk
- TUMOR REGRESSION: Absent
- MARGINS: Extending to inked lateral and deep resection margins
Case 1 Clinical Questions (continued):
What is this patient’s T stage?
What would be your next steps in work up?
- What would be your surgical plan?
- What additional studies (if any) would you order?
- What would be your surgical plan?
CASE 2
Synopsis
- A 68-year-old Male with a history of HTN, HLD, T3bN1aM0 melanoma presents for follow up to discuss next steps in management.
Relevant Work Up To Date:
- Tumor:
- 3.2 mm thick, ulcerated SSM with 5 mitoses, Right mid back
- 3.2 mm thick, ulcerated SSM with 5 mitoses, Right mid back
- Nodal:
- 1/14 nodes positive for melanoma on sentinel lymph node biopsy
- 1/14 nodes positive for melanoma on sentinel lymph node biopsy
- Imaging:
- No evidence of distant disease
- No evidence of distant disease
- Molecular Testing:
- BRAF p.V600E (c.1799T>A): absent
- BRAF p.V600K (c.1798_1799delGTinsAA): present
- INTERPRETATION: POSITIVE for variant in BRAF.
- BRAF p.V600E (c.1799T>A): absent
Case 2 Clinical Questions:
- What is this patient’s pathological stage?
- What are the therapeutic options for this patient?
CASE 3
Synopsis
- A 58-year-old Female with a history of HTN, HLD, presents with a 3 cm ulcerated pigmented lesion on the right thigh. Physical exam reveals a 6 cm right inguinal mass.
Case 3 Clinical Questions:
- What are your next steps in management?
- If regionally metastatic melanoma is confirmed, what management options are available?
CASE 4
Synopsis
- A 49-year-old Male with a history of pulmonary sarcoidosis presents with BRAF V600 mutated melanoma metastatic to the liver and lungs.
Case 4 Clinical Questions:
- What therapeutic strategies are available for this patient?
- Given the patient’s comorbidities, what would be your first-line treatment strategy?
Timeline of FDA-Approved Medications for Melanoma
Overview
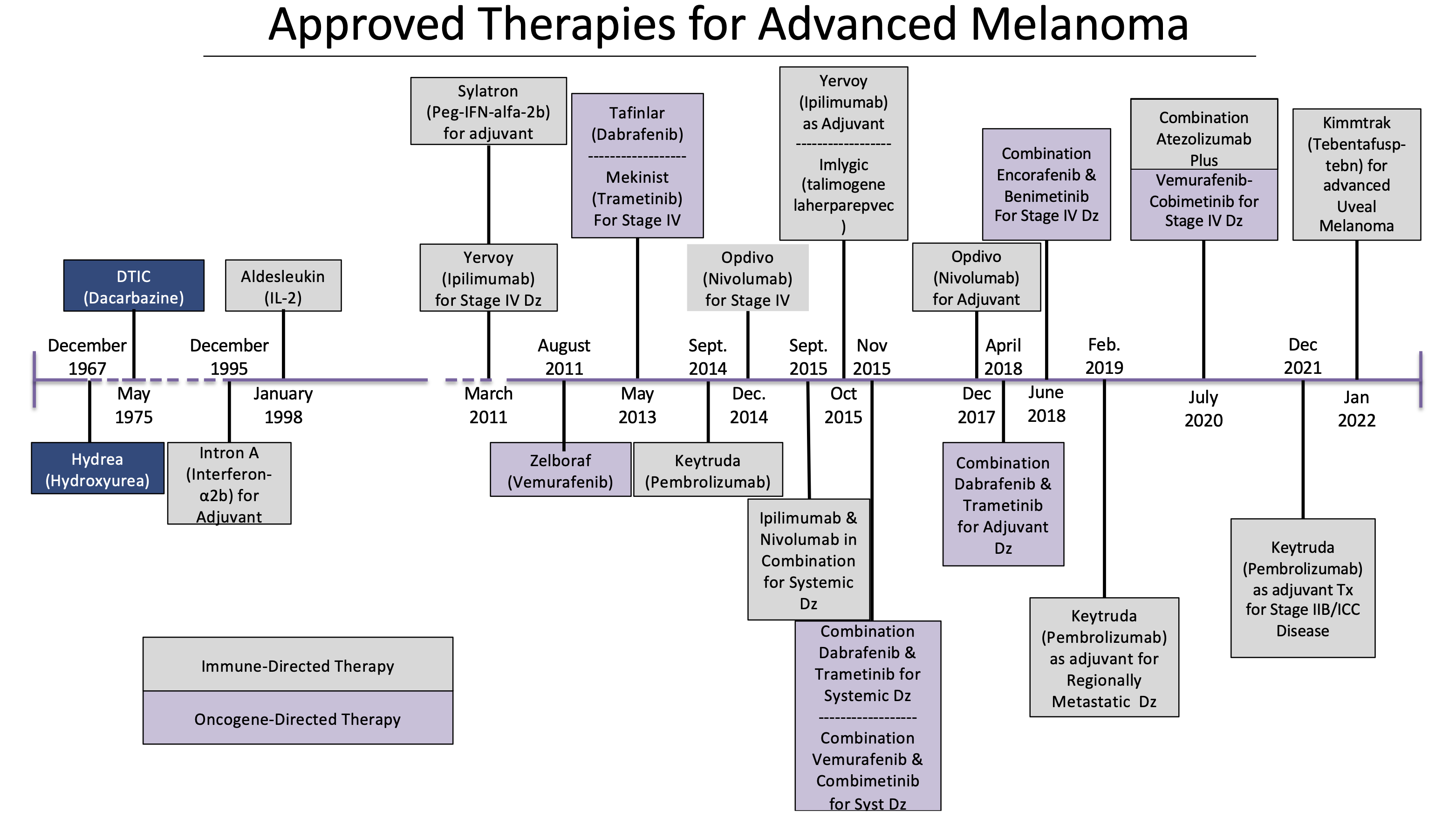
The therapeutic landscape for patients with melanoma has changed significantly over the last 60 years. Advances in both oncogene-targeted therapy and immuno-oncological strategies have improved recurrence-free and overall survival. Below is a data visualization of the therapies approved to treat high risk and advanced disease.
Background on Oncogene-Targeted Strategies
Overview
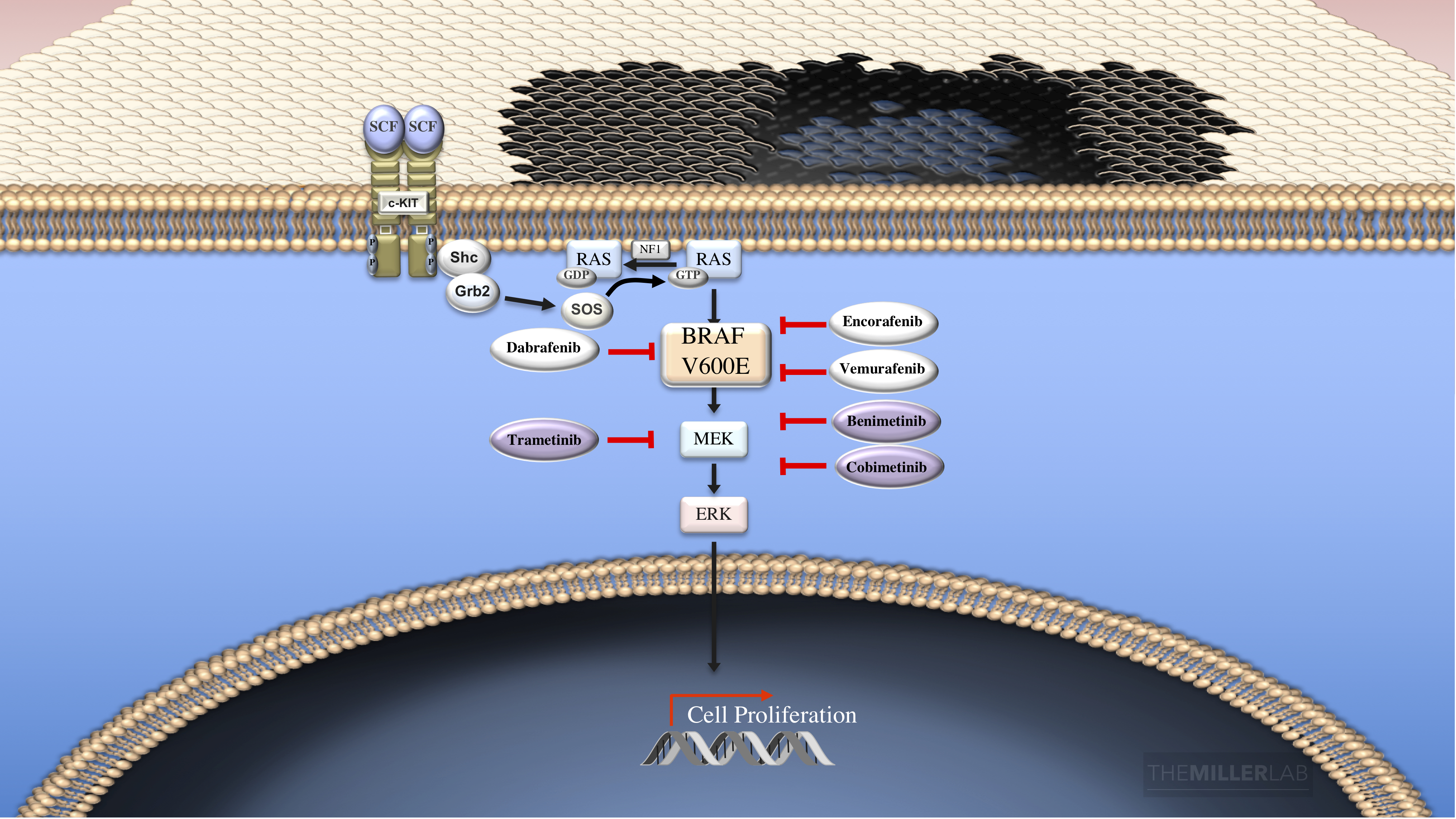
As stated above, the recognition that gain-of-function mutations in BRAF occur in nearly half of melanomas lead the way to drug development programs targeting BRAF and the downstream effector MEK. At this time, three pairs of BRAF-MEK combinations are FDA approved for patients with BRAF mutant melanoma (Dabrafenib-Trametinib; Vemurafenib-Cobimetinib; Encorafenib-Benimetinib). Please see the figure below for more details.
Background on Immuno-Oncological Strategies
Overview
Immune-directed therapies (cytokine therapy, immune-checkpoint blockade, oncolytic viral therapy and cellular therapy) have revolutionized the care of patients with high-risk and advanced melanoma. We will discuss the application of these strategies in greater detail during the lecture, but we’ll provide a very basic primer of T-cell activation and immune checkpoint blockade below.
T-Cell Activation
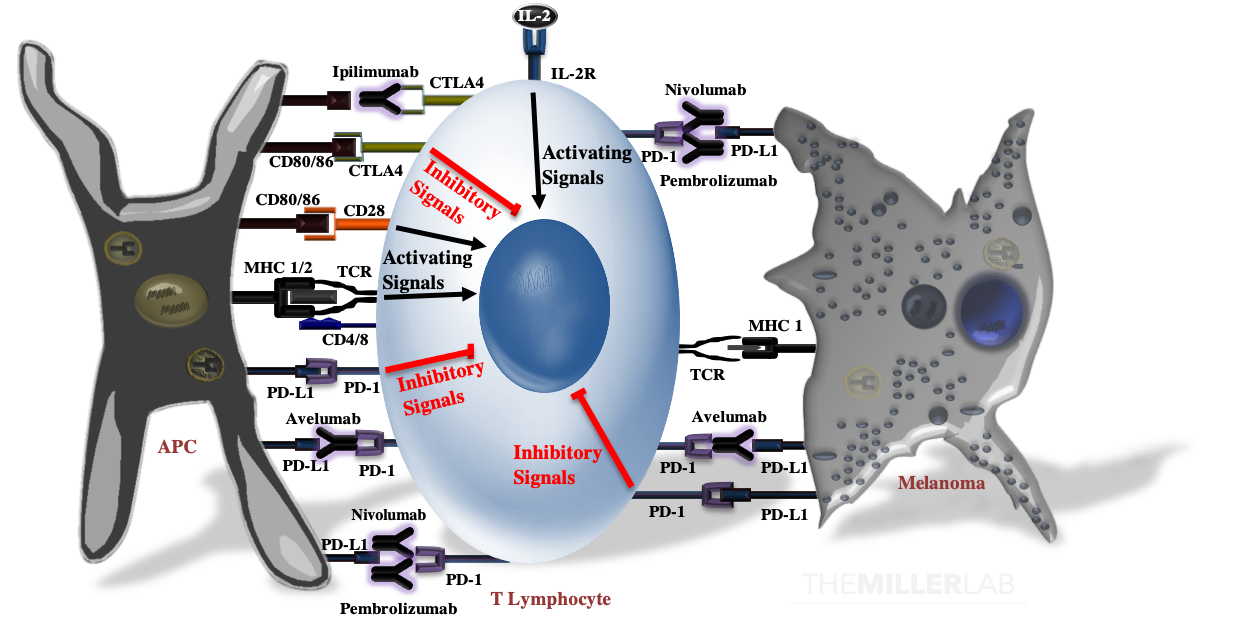
T-cell activation results from the summation of several activating signals (Below Figure). These include stimulation of the T-cell receptor (TCR) by an antigen complexed with the major histocompatibility complex (MHC) and CD28 by CD80 or CD86 (CD80/86). Of note, MHC class I molecules activate CD8+-T cells, while MHC class II molecules are recognized by CD4+-T cells. T-cell activation and function are tightly calibrated by regulatory mechanisms to prevent autoimmunity and excessive tissue inflammation. T-cell activation is negatively regulated by CTLA4, which not only competes with CD28 for CD80 and CD86, but can also transmit inhibitory signals that attenuate activation. In peripheral tissues, ligation of the program death-1 (PD-1) receptor by PD-L1 inhibits effector functions of T-cells. PD-L1 is expressed on both antigen-presenting cells, such as macrophages, and malignant melanocytes.
Immune Checkpoint Blockade
Monoclonal antibodies directed against CTLA-4 (e.g. ipilimumab), PD-1 (e.g. nivolumab and pembrolizumab) and PD-L1 (e.g. avelumab and atezolizumab; of note, neither of these last two are approved for melanoma, but they have labeled inidcations for other malignancies) can overcome intrinsic immune checkpoints by licensing tumor-specific T cells.2
References
Bennett, D. C. (2008). How to make a melanoma: what do we know of the primary clonal events? Pigment Cell Melanoma Res 21, 27-38.
Davies, H., Bignell, G. R., et al. (2002). Mutations of the BRAF gene in human cancer. Nature 417, 949-54.
Funayama, R., and Ishikawa, F. (2007). Cellular senescence and chromatin structure. Chromosoma 116, 431-40.
Malumbres, M., and Barbacid, M. (2001). To cycle or not to cycle: a critical decision in cancer. Nat Rev Cancer 1, 222-31.
Malumbres, M., and Barbacid, M. (2009). Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 9, 153-66.
Malumbres, M., Sotillo, R., et al. (2004). Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell 118, 493-504.
Miller DM, Flaherty KT. Cyclin-dependent kinases as therapeutic targets in melanoma. Pigment Cell Melanoma Res. 2014;27(3):351-365.
Miller DM, Flaherty KT, Tsao H. Current status and future directions of molecularly targeted therapies and immunotherapies for melanoma. Semin Cutan Med Surg. 2014;33(2):60-67.
Pardee, A. B. (1974). A restriction point for control of normal animal cell proliferation. Proc Natl Acad Sci U S A 71, 1286-90.
Serrano, M., Lee, H., et al. (1996). Role of the INK4a locus in tumor suppression and cell mortality. Cell 85, 27-37.
Serrano, M., Lin, A. W., Mccurrach, M. E., Beach, D., and Lowe, S. W. (1997). Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88, 593-602.
Wang, W., Chen, J. X., et al. (2002). Sequential activation of the MEK-extracellular signal-regulated kinase and MKK3/6-p38 mitogen-activated protein kinase pathways mediates oncogenic ras-induced premature senescence. Mol Cell Biol 22, 3389-403.
Footnotes
- This paragraph was adapted from: Miller DM, Flaherty KT. Cyclin-dependent kinases as therapeutic targets in melanoma. Pigment Cell Melanoma Res. 2014;27(3):351-365
- This paragraph was adapted from: Miller DM, Flaherty KT, Tsao H. Current status and future directions of molecularly targeted therapies and immunotherapies for melanoma. Semin Cutan Med Surg. 2014;33(2):60-67
This site represents our opinions only. See our full Disclaimer and Terms of Use Agreement