Optimizing Real-World Data Collection: Clinical Genomics
2020
RWD
Abstract
Facilitate capture of real-world next-generation genomic data
Objectives:
- Clinical Real-World Data (RWD) captured outside the context of clinical trials from electronic health or medical records (EHR/EMR) is a rich source of information
- Transforming this unstructured digital repository of treatments, outcomes, and the longitudinal patient experience into highly structured data poses significant challenges for real-world setting data capture and analysis
- Therefore, strategies to facilitate the collection of RWD are sorely needed
- Of note, data collection from EHRs should only be conducted under a protocol approved by the respective Institutional Review Board (IRB).
Clinico-Genomics Databases
- Innovation in the treatment of cancer begins with a deep understanding of tumor biology
- Clinico-Genomic Patient Registries allow for associations between tumor genomics and patient characteristics with clinical outcomes to learn from real-world data.
- Linking real-world clinical outcomes to genomics data provides powerful study cohorts. This enables data-driven interventions in precision medicine.
- We describe an approach to facilitate capture of the clinical next-generation genomic data from EHRs to complement real-world clinico-genomic databases of patients with cancer.
- In this post, we will outline methodology and design, utilizing REDCap to capture the genomic data from clinical tumor molecular profiling that has increasingly guided oncology treatment and care. Capturing the data in REDCap allows for unstructured-to-structured transformation and absolute ease for down stream analysis. We briefly describe how to curate a Master gene list for capturing the data.
Overview of REDCap
- REDCap is a user-friendly, web-based research electronic data capture platform utilized by researchers worldwide to collect structured data for statistical analysis.1,2
- It is a powerful, secure, and HIPAA compliant database building solution created by Vanderbilt University in 2004 that allows development and deployment of subject facing surveys as well as forms for data capture by data entry personnel
- Our lab has adopted REDCap as the data collection platform for our rare cutaneous tumor patient registries. We have additionally deployed modules built in REDCap to streamline and analyze operations in the multidisciplinary clinic setting
Capturing Tumor Sequencing/Genomics Data:
Key Points
- In this post we provide tips and recommendations to provide ease for the design of a genomics data capture instrument for use in a REDCap clinico-genomics database
- Recommendations for design to avoid downstream pitfalls on the backend.
- After this monograph, you will:
- Know what aspects to consider in the design and deployment of a Genomics based instrument for capturing clincially acquired next generation sequencing (NGS) results from the EHR into the registry.
- Understand how to routinely update newly added genes/features of targeted panels for sequencing as they expand to cover more genes that prove to be relevant overtime.
- Skill Level: Intermediate Knowledge of REDCap Insrument Database design and basic data wrangling in R is required.
Aim 1: Accomodating Several Sequenced Tissues per record
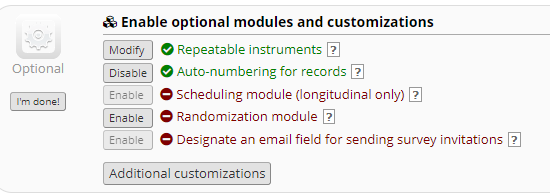
- One record may have several tissues sequenced, for example a primary tumor and then a metastases. So we will enable the REDCAp repeating instruments function for the Genomics Capture by enabling Repeatable instruments in the Project Setup..
- In the event that more than one instrument is created per record for different tissue samples, the ability to label each tissue by the specific lesion is important. Providing a Tissue Lesion Identifier is essential. However, throughout all the instruments in any clinico-genomics database, consistency aids back end analysis. It is useful to have the same lesion identified with the same identifier or tag in every instrument instance.
- For example, if a lesion on the upper right extremity has an instance in the Pathology instrument and the Genomics instrument, it is most useful to have the same lesion labeled the same way for correct association on the back end and to facilitate analysis.
- One way to promote accuracy and consistency with exact nomenclature is to utilize REDCap’s piping feature and pipe the labels given in one instrument into other instruments.
- This helps to ensure exact duplicated tag names by the abstractor.
Aim 2: Ensure Record Capture Completeness
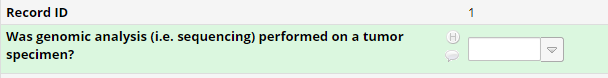
- It is unlikely every record will have tissue that undergoes institutional next generation sequencing. Therefore, it is important to add a “Yes/No” field to indicate if genomic analysis was performed on a tumor specimen.
- This allows the abstractor to answer “NO” after thoroughly searching the EHR chart. A”NO” option at the current time allows for assurance the record has been searched and there is not missing genomics data yet to be entered.
- This is beneficial for the abstractors performing a second pass abstraction, as well as for the database manager and the QC aspect of the database pipeline.
Aim 3: Unique identifiers
- Create dropdown fields to allow designation of the type of tissue sequenced and the date it was performed.
- This will allow for unique identifiers for each sequencing date, for each tissue specimen within each record.
Aim 4: Targeted Assay version-year
- Create a dropdown menu that allows for entering the type of targeted NGS assay performed.
- This is essential since different platforms have different coverage and sequence different gene panels.
- Furthermore including information on the assay reveals which genes were wildtype as opposed to never sequenced.
- Most sequencing platforms report the altered genes, not every gene on the panel that came back wildtype.
- The version/year of the sequencing platform is also important since different versions of the same platform sequence different genes. For example, over the last five years, some targeted NGS-assays have added over 200 new clincially relevant genes to their panel.
- The full list of genes included in each platform should be maintained for back end use.
Aim 5: Which gene options to include?
Once the repeatable genomics instrument and the identifying specimem dropdown fields are created, it’s essential to decide which genes will be pre-populated in drop down menus as options to enter genomics data abstracted from the EHR.
As previously mentioned, after identifying the main NGS platforms typically used, curate a total gene list of all the genes tested in all the panels. This will serve as the Master Gene List. Obviously, many of the genes will overlap among the different assays or use different nomenclature. R software can be used to curate one final list.
For example, let’s create a simplified list for demonstration purposes.
Load Libraries
library(tidyverse)
library(reshape2)
library(kableExtra)
library(data.table)
library(kableExtra)
library(dplyr, warn.conflicts=FALSE) # Useful for manipulating the dataframes
library(knitr)Let’s create a data set of assays and associated genes
- This is an overly simplified number of assays with reduced lists for demonstration
- In reality over ten different assays may be used each with 100-800 different genes
Master_Gene_List = data.frame (ASSAY= c('Assay A', 'Assay B', 'Assay C', 'Assay D'), Gene1= c("EGFR", "EGFR", "ALK", "BRAF"), Gene2= c("ALK", "PIK3CA", "ARID1A", "TP53"), Gene3= c("TP53", "B-raf", "TERT", "SMARCF1")) Let’s view the list of assays and associated genes
kable(Master_Gene_List) %>%
kable_styling(bootstrap_options = "striped", full_width = F)| ASSAY | Gene1 | Gene2 | Gene3 |
|---|---|---|---|
| Assay A | EGFR | ALK | TP53 |
| Assay B | EGFR | PIK3CA | B-raf |
| Assay C | ALK | ARID1A | TERT |
| Assay D | BRAF | TP53 | SMARCF1 |
Notice that different assays may contain the same exact genes. Furthermore, different assays may refer to the same genes in their respective panels but with different nomenclature or symbols.
Therefore, some tidying of the master list is needed. Once we have a finalized list of genes, it can be used in the dropdown menu in the REDCap genomics instrument.
Alternatively, each gene list can be kept as is and once a user clicks on the assay used for the sequencing, branching logic can be implemented and only allow dropdown menus with those assay-specific genes
However, we have found it easier to update the instrument with new genes using one master list, as opposed to new individual assay lists
- Furthermore, QC is more amenable to gene errors on one list as opposed to branching logic
- For example, if the assay type or version was incorrect and upon QC switched to the correct assay type, the rest of the branching logic dependent data entered will be dependent on the wrong assay and deleted
- These gene lists are long and it is easier to maintain one main gene list than separate ones for separate assays
- For example, if the assay type or version was incorrect and upon QC switched to the correct assay type, the rest of the branching logic dependent data entered will be dependent on the wrong assay and deleted
- Furthermore, QC is more amenable to gene errors on one list as opposed to branching logic
Note that several paths work and the reader is urged to test what works best for their system.
If the user chooses to have the assay specific list, then stop here. Once an assay is chosen, the assay-specific dropdown menu should branch off and provide the abstractor with the specific assay associated gene list. These genes should be the only ones available for that NGS-specific assay.
For example, if the abstractor chooses “Assay A,” three fields with dropdown menus for indels, fusions, and SNVs will only include the genes associated with that assay. Note in this example, that would be ‘EGFR’, ‘ALK’, and ‘TP53’
- This Assay Dependent Gene List method will still require genes with different names/symbols to be wrangled and replaced with one common preferred symbol/name for faciliation and ease of analysis.
Below, we outline how to create one master list, remove duplications, and ease data abstractor gene choices. Remember, with updated panels, some adopt new nomenclature of the genes instead of alias/archaic names
Thus, it is important UX/UI is considered to ensure ease as well as accuracy in data entry.
Creating one Master Gene List
- For one master list, we will next remove the duplicated genes.
- This Master list will be used in the dropdown menu for indels, fusions, and SNVs for the abstractor to enter genomics data, independent of the assay selection.
- Since the Master Gene List is independent of the assay, we will melt the datatable and remove duplications.
Genes<- melt (Master_Gene_List %>% select("ASSAY", "Gene1", "Gene2", "Gene3"), id.var="ASSAY")Let’s view the melted table
kable(Genes) %>%
kable_styling(bootstrap_options = "striped", full_width = F) | ASSAY | variable | value |
|---|---|---|
| Assay A | Gene1 | EGFR |
| Assay B | Gene1 | EGFR |
| Assay C | Gene1 | ALK |
| Assay D | Gene1 | BRAF |
| Assay A | Gene2 | ALK |
| Assay B | Gene2 | PIK3CA |
| Assay C | Gene2 | ARID1A |
| Assay D | Gene2 | TP53 |
| Assay A | Gene3 | TP53 |
| Assay B | Gene3 | B-raf |
| Assay C | Gene3 | TERT |
| Assay D | Gene3 | SMARCF1 |
Now lets keep only distinct genes in the ‘value’ column and view this new table
kable(Genes %>% distinct(value, .keep_all= TRUE)) %>%
kable_styling(bootstrap_options = "striped", full_width = F) | ASSAY | variable | value |
|---|---|---|
| Assay A | Gene1 | EGFR |
| Assay C | Gene1 | ALK |
| Assay D | Gene1 | BRAF |
| Assay B | Gene2 | PIK3CA |
| Assay C | Gene2 | ARID1A |
| Assay D | Gene2 | TP53 |
| Assay B | Gene3 | B-raf |
| Assay C | Gene3 | TERT |
| Assay D | Gene3 | SMARCF1 |
- As you can see, the duplicated EGFR and TP53 have been removed. Again this Master Gene List is independent of the Assay so we will only be adding the ‘value’ column to the dropdown menu list in REDCap indels, SNVs and fusion genomics field.
- Note that “BRAF” and “B-raf” are the same gene but appear with different nomenclature/symbols. We have found that abstractors are not necessarily familiar with all annotations/symbols of every gene. A simple solution is to include the other options for the gene symbol so that if the abstractor starts to type “BR” in the field with the drop down menu or “b-”, only one correct option in the drop down menu appears in REDCap
- So, lets replace ‘BRAF’ with “BRAF (B-raf)” to facilitate the data abstractor choosing the same gene whether entering ‘B-raf’ mutations from Assay C or “BRAF” mutations detected from Assay D.
Genes$value <- replace(Genes$value, Genes$value == "B-raf", "BRAF (B-raf)")- Let’s drop the BRAF alone value
Genes<-subset(Genes, value != "BRAF" )- Let’s view the new table
kable(Genes %>% distinct(value, .keep_all= TRUE)) %>%
kable_styling(bootstrap_options = "striped", full_width = F) | ASSAY | variable | value |
|---|---|---|
| Assay A | Gene1 | EGFR |
| Assay C | Gene1 | ALK |
| Assay B | Gene2 | PIK3CA |
| Assay C | Gene2 | ARID1A |
| Assay D | Gene2 | TP53 |
| Assay B | Gene3 | BRAF (B-raf) |
| Assay C | Gene3 | TERT |
| Assay D | Gene3 | SMARCF1 |
- Note that adjusting this list is simple when only a few rows exist. We could have used “ifelse” statements in R or replaced both with “BRAF (B-raf)” with partial matches and then removed duplications
- However, with a list of thousands of genes, this is not trivial. Also, note that ‘Partial matches’ may alter other unanticipated gene names when dealing with larger data set
- Furthermore, even in this small gene list, only a curator familiar with gene symbols would have noticed that ‘ARID1A’ has an alias, ‘SMARCF1’.
- However, with a list of thousands of genes, this is not trivial. Also, note that ‘Partial matches’ may alter other unanticipated gene names when dealing with larger data set
#Thus we can adjust this as before.
Genes$value <- replace(Genes$value, Genes$value == "SMARCF1", "ARID1A (SMARCF)")
#Let's drop the ARID1A alone value
Genes1<-subset(Genes, value != "ARID1A" )
#Let's view the new table
kable(Genes1 %>% distinct(value, .keep_all= TRUE)) %>%
kable_styling(bootstrap_options = "striped", full_width = F) | ASSAY | variable | value |
|---|---|---|
| Assay A | Gene1 | EGFR |
| Assay C | Gene1 | ALK |
| Assay B | Gene2 | PIK3CA |
| Assay D | Gene2 | TP53 |
| Assay B | Gene3 | BRAF (B-raf) |
| Assay C | Gene3 | TERT |
| Assay D | Gene3 | ARID1A (SMARCF) |
HGNC Human Gene Nomenclature and Aliases
- A much more practical approach is to download an annoted gene list with symbols and aliases.
- Use the downloaded list to curate the final Master Gene List.
- The downloaded list can be used for search and replace or as a lookup table.
- Furthermore, always provide an “other” option with a branching free text field for unanticipated genes that may need to be collected before updates are completed.
- We prefer downloading sources located on the HGNC (HUGO Gene Nomenclature Committee) site, which provides several downloadable lists of approved gene human gene nomenclature.